
Sindrome di Gianotti-Crosti
Dott. Luciano Schiazza
Specialista in Dermatologia e Venereologia
Specialista in Leprologia e Dermatologia Tropicale
cell 335.655.97.70
www.lucianoschiazza.it

Prima di descrivere la sindrome indicata nel titolo, desidero ricordare il Prof. Ferdinando Gianotti, avendo avuto il privilegio di conoscerlo sia nel suo ruolo istituzionale sia in occasione meno formale quali erano le riunioni del Tigliole Skin Club.
A Lui si deve, nel 1954, la creazione del primo reparto di Dermatologia Pediatrica Italiano e, probabilmente, in Europa.
Risale al 1953 la sua prima osservazione di un bambino affetto da una eruzione monomorfa eritematopapulosa, localizzata sulla superficie estensoria degli arti superiori e inferiori, accompagnata da linfoadenopatia generalizzata. Egli non riusciva a inquadrarla in una patologia già conosciuta, sospettando trattarsi di qualcosa di nuovo e ipotizzandone la natura infettiva.
Nella pubblicazione del primo caso (1955), sottolineava come la dermatosi differiva dalle altre patologie virali o batteriche in quanto la febbre e le manifestazione sistemiche erano minime nel suo caso e che la cute interessata dall’eruzione erano gli arti (acrale), con un tempo di permanenza variabile tra i 25 ed i 45 giorni, periodo ben più lungo dei conosciuti esantemi infettivi. Il prof Gianotti scriveva “credo che questa malattia sia associata ad un virus, poiché il paziente con la dermatosi acrale e altri 5 bambini che erano stati in contatto con lui, avevano esami ematologici con le stesse caratteristiche –leucocitosi sino a 11.500/mm cubici sino al 56% di monociti e cellule di Turk. Sospetto una unica, sconosciuta origine virale che in alcuni pazienti determina una eruzione cutanea”.
Le cellule di Turk sono delle grosse cellule mononucleate con i caratteri di un linfocita atipico e di una plasmacellula con nucleo eccentrico e citoplasma basofilo.
I colleghi ed il Direttore della Clinica Dermatologica di Milano, Prof Agostino Crosti, inizialmente erano scettici sulle sue conclusioni “Gianotti, ti té i ghe la mania del virus”. Tuttavia la identificazione di altri 8 casi nell’anno seguente convinsero il Prof Crosti dell’esistenza della malattia e dell’importanza della scoperta del Dott. Gianotti.
Furono pubblicati due lavori sotto il titolo “dermatosi infantile eruttiva acroposta di probabile origine virale” che, come tradizione dei tempi, metteva come primo autore il Direttore della Clinica, da cui l’iniziale identificazione come sindrome di Crosti-Gianotti successivamente poi variata in sindrome di Gianotti-Crosti (SGC).

Solo nel 1970 fu confermata la relazione dermatosi acroposta e virus, in particolare l’antigene Australia, in seguito identificato con antigeni di superficie dell’epatite B (HBsAg), la cui positività conferma la potenzialità infettiva del paziente.
Successivamente la scoperta di altri casi negativi per il virus dell’epatite indicarono la correlazione infettiva della sindrome con svariati altri virus: virus Epstein-Barr (EBV) virus epatite A; cytomegalovirus (CMV); herpesvirus umano 6 e 7 (HHV6-7); coxsackievirus A16, B4, B5; rotavirus; parvovirus B19; virus sinciziale respiratorio; virus della parotite; virus parainfluenzale tipo 1 e 2. Seppur più raramente anche batteri furono chiamati in causa: Bartonella henselae, streptococco β emolitico, Mycoplasma pneumoniae, Borrelia Burgdorferi.
Oltre alla forma maculo-papulosa furono osservati casi in cui l’eruzione aveva carattere papulo-vescicoloso, inducendo a distinguere le due eruzioni in una forma dipendente dal virus HBV e l’altra da virus non HBV.
L’osservazione retrospettiva di oltre 300 casi da parte del Prof Caputo Direttore della Clinica Dermatologia dell’Università di Milano e di altri colleghi dermatologi, portò alla conclusione che non vi erano differenze cliniche tra le forme indotte o no dal virus HBV e che le variazioni del pattern cutaneo erano probabilmente determinate da altri fattori quali l’età, lo stato generale di salute e lo stato immunitario del paziente. Pertanto la malattia doveva essere definita come sindrome di Gianotti-Crosti, comprensiva e diagnostica per entrambe le variazioni eruttive cutanee.
I pazienti affetti da GCS hanno età compresa tra i 3 mesi ed i 15 anni, con maggiore incidenza tra 1 e 6 anni e picco di età ai 4 anni. Anche gli adulti possono esserne affetti con esclusività per il sesso femminile.
Le variazioni stagionali di comparsa seguono l’incidenza delle infezioni virali scatenanti.
Clinicamente l’eruzione cutanea esordisce improvvisamente, talvolta preceduta da sintomi quali faringite, infezione delle vie aeree superiori, diarrea.
Compaiono numerose papule o papulo-vescicole monomorfe di colore rosa o rosso-bruno, lievemente pruriginose, del diametro variabile tra 1 e 5 mm con la tendenza alla confluenza.

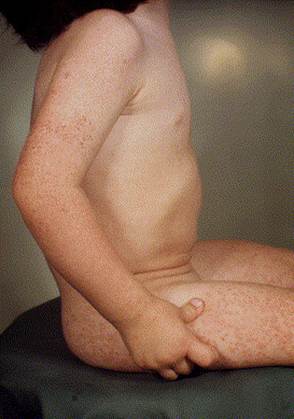
Si localizzano in maniera simmetrica sulle guance, sulla superficie estensoria degli arti superiori e inferiori ed alle natiche. Il tronco, le ginocchia ed i gomiti sono comunemente risparmiati se non occasionalmente con pochi e rari elementi. Le mucose non sono interessate.
Dal punto di vista generale, può comparire sensazione di malessere, febbricola o diarrea. In un terzo circa dei pazienti è presente ingrossamento dei linfonodi cervicali, ascellari o inguinali.
Gli aspetti istologici per quanto evidenti non sono specifici.

La variante vescicolosa presenta una lieve acantosi con diffusa spongiosi e vescicole, al cui interno si osservano cellule di Langerhans e occasionalmente linfociti. Nel derma si osserva un infiltrato linfocitario, come in molte dermatosi infiammatorie.

La variante non vescicolosa, l’epidermide evidenzia modesta acantosi, ipercheratosi paracheratosi focale e spongiosi (nelle forme di più vecchia data). Nel derma si osserva un infiltrato perivascolare soprattutto linfocitario (con qualche istiocita).
Comunemente le manifestazioni cutanee permettono di fare diagnosi a prima vista. Tuttavia la GCS può assumere aspetti atipici, soprattutto all’esordio o nelle fasi di remissione.
Si pone quindi la diagnosi differenziale con:
- Lichen planus, caratterizzato da prurito intenso, coinvolgimento preferenziale della superficie volare dei polsi, colore violetto chiaro, raro interessamento delle guance, frequente delle mucose.
- Reazioni lichenoidi da farmaci, poco frequenti nei bambini e localizzate soprattuto al tronco.
- Eritema multiforme, nelle sue fasi iniziali, spesso papulose, solitamente scatenate da infezioni ricorrente di Herpes simplex virus.
- Scabbia, talvolta con manifestazioni prevalenti agli arti. Nella scabbia però il prurito è intenso con accentuazione notturna (nella GCS se presente non è significativo), sono presente escoriazioni (assenti nella GCS), sono interessate le areole mammarie, il glande, l’ombelico ( non interessate nella GCS).
- Orticaria papulosa (detta anche prurigo simplex acuta o strofulo). In questo caso le lesioni papulose pruriginose sono raggruppate sulla superficie estensoria degli arti e spesso anche al tronco. Essendo la causa riferibile ad artropodi la stagionalità è limitata alla primavera/estate e la loro durata è limitata a pochi giorni.
Il decorso della GCS è, per quanto lungo, benigno (nel 99% dei casi), con risoluzione senza esiti delle lesioni. La linfoadenopatia può durare parecchi mesi come l’epatosplenomegalia, se presente.
La natura autorisolutiva della GCS unitamente alla scarsità sintomatologica non inducono spesso ad un supporto terapeutico ma piuttosto la rassicurazione dei genitori. Se necessaria, in caso di prurito, antistaminici per via orale e/o topici cortisonici di media potenza. In casi particolarmente gravi è stata intrapresa terapia cortisonica per via sistemica.
